Matthew Smith

Matthew Smith
Member of Magdelene College
PhD student in Prof Payne's group
Office: 538 Mott Bld
Phone: +44(0)1223 3 37216
Email: mjs281 @ cam.ac.uk
TCM Group, Cavendish Laboratory
19 JJ Thomson Avenue,
Cambridge, CB3 0HE UK.
Research
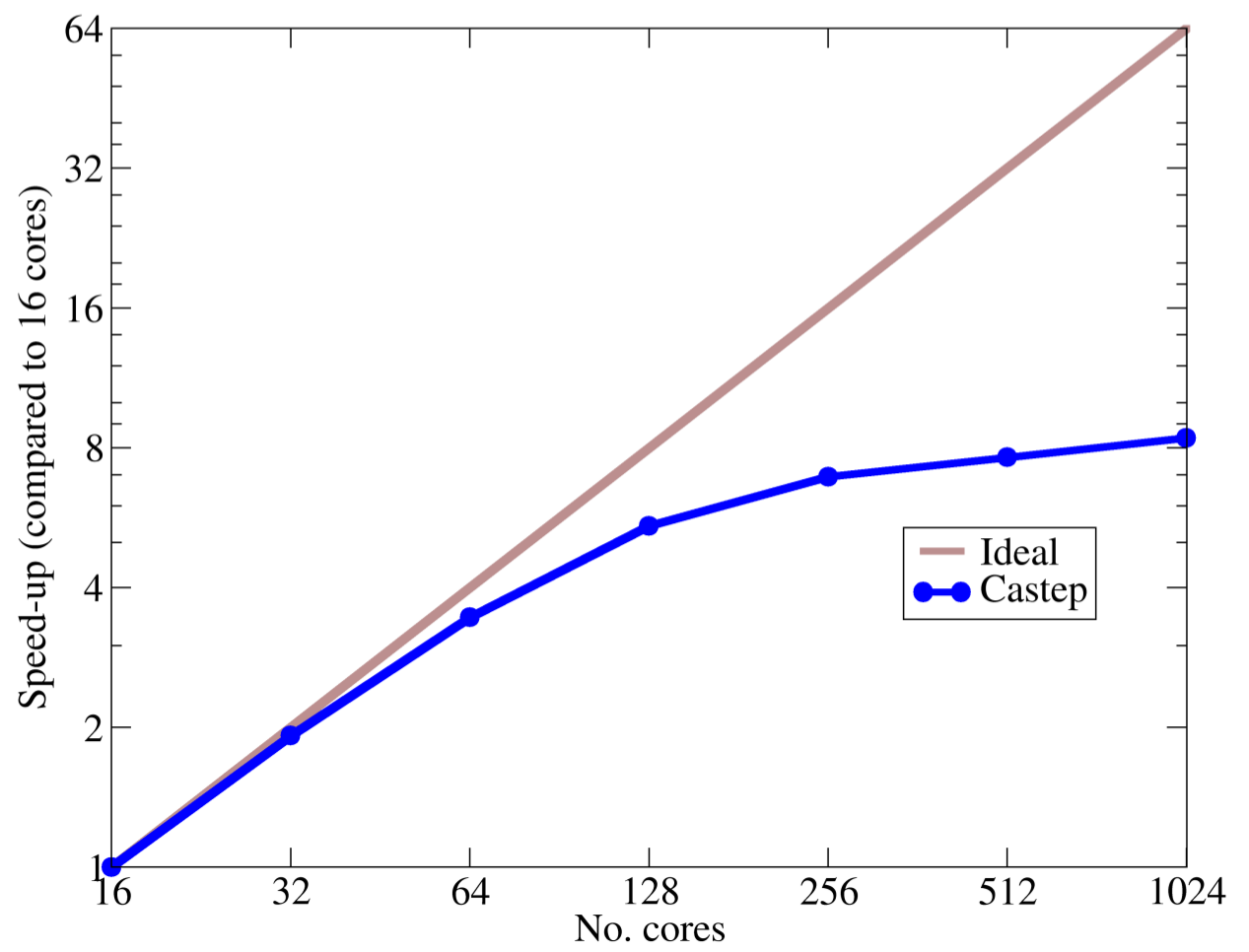
Density functional theory provides a framework for efficient and accurate computational modelling of materials from first principles. It has been tremendously successful at describing the properties of atoms, molecules, nanostructures, and solids, and its success has been possible because of the development of software implementations. One such implementation is CASTEP (http://www.castep.org), a software pakcage developed jointly by researchers at the Universities of Cambridge, Durham, Oxford, Royal Holloway and York. Although much of CASTEP is highly optimised for parallel calculations on conventional high performance computing architectures, communication costs incurred when performing fast Fourier transforms currently cause a significant divergence from the ideal of linear parallel scaling with number of processes. Hence, it is intended to minimise these communications by implementing a domain decomposition algorithm capable of providing the optimal decomposition for any combination of simulation size and number of processes.
It is also intended to develop CASTEP in order to take advantage of recent hardware developments. These have seen a move towards large numbers of low-power cores, such as those found in general purpose graphics processing units. The theoretical peak performance of these emerging technologies is enormous, but exploiting their unusual architecture effectively requires a re-imagining of many of CASTEP's key operations and the implementation of new software paradigms (e.g. OpenACC). Exploiting these cutting-edge technologies effectively will dramatically increase the performance of the software and enhance its effectiveness in a number of key materials modelling research areas. These include high-throughput calculations, such as those involving global geometry optimisations (see, for example, AIRSS), molecular dynamics, as might be used to compute finite-temperature band-gaps, and investigations using density functional perturbation theory for phonons. It is the last of these areas which is of particlar interest, as it is intended that the newly optimised CASTEP will immediately be used to investigate the thermo-electric properties of nanophononic metamaterials. Models of such systems are beyond the present capabilities of CASTEP, as the thousands of atoms typically involved push the software beyond the limits of its parallel scaling. Hence, the development of the software will open up its application to new areas of scientific interest.
In Plain English
My First Heading
The vast majority of conventional high performance computers today are really clusters of processors. Each processor in a cluster is, more or less, similar to that found in a typical desktop machine. High performance is achieved by having each processor perform tasks independently of all others, and hence allowing a number of tasks to be performed simultaneously. This is usually what is meant by computing `in parallel'. In order for tasks to be performed in parallel, it is often necessary that processors share information with one another from time to time - a process known as `communicating'. Communications are typically much slower than computations, and therefore the optimal performance of a piece of software running in parallel on a conventional high performance cluster can only be achieved if the program is written in such a way as to minimise communications.
A complimentary way of achieving optimal performance is to look beyond conventional machines, and exploit the computing power offered by novel techonolgies. One of the most popular and promising of the emerging hardware paradigms is general purpose graphics programming units. Each of these units is, in effect, its own self-contained computing cluster - thousands of low-powered processors capable of performing large numbers of similar tasks in parallel. The architecture of these units is fundamentally different from that of conventional processors and hence, in order to exploit their computing power, it is necessary to re-write or even re-conceive algorithms designed to be run on conventional clusters.
Featured Publications
Pending.